
The editors and current author would like to thank and acknowledge the significant contribution of the previous author of this chapter from the 2004 first edition, Dr. Steven C. Crook. This current second edition chapter is a revision and update of the original author’s work.
Case 1
A 3-year-old male presents to the ER with a gross deformity of his left upper arm. His mother reports that her son "slipped while taking a bath" and struck his arm on the side of the tub. He has a history of a previous right femur fracture that resulted from a fall down the stairs one year ago. In addition to the gross deformity to the left humerus, his physical exam is notable for bluish sclera and no other signs of trauma (no bruising or scars). A skeletal survey is ordered by the ER physician, who thought that the injury did not fit the history provided by the parent. The plain films reveal a comminuted fracture in the middle of the left humerus, and signs of multiple healed rib fractures in addition to the healed right femur fracture. The ER physician notifies child protective services (CPS) as required by law. He is later referred to a geneticist who makes the diagnosis of osteogenesis imperfecta.
Case 2
A 16-year-old male presents to the ER with a chief complaint of a sudden onset of left chest pain. The pain was non-radiating on the left chest, sharp and 8/10 severity. It is associated with shortness of breath. It occurred while he was watching TV at home an hour ago. He denies any trauma to his chest and any cough or other respiratory symptoms prior to the onset of pain. Past medical history is unremarkable and takes no medications. He denies any cigarette smoking or recreational drug use. On physical examination, he appears to be a tall and slender individual who wears glasses. His temperature is normal, pulse 110/min, blood pressure 137/88, and respiratory rate 28/min, oxygen saturation 97%. The pulmonary exam reveals diminished breath sounds on the left but normal on the right. Cardiac auscultation reveals regular rhythm with no murmurs or gallop. EKG is normal, troponin I and CK-MB are negative. The chest radiograph shows a 60% left pneumothorax, without effusion or pulmonary lesions. Chest tube is placed and he is placed on oxygen. His pneumothorax resolved after 2 days of hospitalization. He is later referred to a geneticist to evaluate him for a hereditary connective tissue disorder.
Connective tissue disorders are inherited disorders of the proteins that comprise various connective tissues such as collagen and elastin. Because these molecules are abundant throughout the body, multiple organ systems are often involved in connective tissue disorders, particularly the skin, heart, eyes, bones and vasculature. The clinical course of these disorders can range widely, from near-normalcy to severe complications. Clinicians should focus on recognizing the potentially life threatening presentations of diseases and differentiating subtle presentations from more common diagnoses. The common inherited connective tissue disorders discussed herein include osteogenesis imperfecta, Marfan syndrome, and Ehlers-Danlos syndromes.
Osteogenesis imperfecta (OI) is a group of disorders often defined by recurrent fractures with minimal trauma, low bone mass and skeletal fragility. OI is also known as "brittle bone disease." Some of the other key features include short stature, blue sclerae, dental abnormalities, and hearing loss. The severity ranges from perinatal lethality to nearly asymptomatic individuals with a mild predisposition to fractures. Hearing loss may be present at birth, develop in childhood, or may never occur. The color of the sclera ranges from a striking dark blue to normal white. OI does not affect cognitive ability. More than 90% of individuals with OI have mutations in the genes COL1A1 or COL1A2 which encode alpha 1 and alpha 2 chains of type I collagen, the main structural protein in bones, ligaments, and tendons. In recent years, mutations in genes encoding proteins involved in the complex post-translational modification of type 1 collagen or in the genetic regulation of bone matrix homeostasis have been identified in association with the other rare OI phenotypes. OI is classified into different types on the basis of the mode of inheritance, clinical presentation, and radiographic findings. OI was originally classified into types I to IV. OI type I to IV exhibit autosomal dominant inheritance and are related to mutations in COL1A1 or COL1A2. In type II, affected infants are either stillborn or die in the early neonatal period as a result of extreme skeletal fragility and pulmonary hypoplasia. The clinical and molecular classification has now expanded to eleven OI phenotypes, many with autosomal recessive pattern of transmission. These classifications of OI types are helpful in prognosis, management, family counseling, and prenatal diagnosis.
The diagnosis is based on family history, history of fractures, characteristic physical findings, and radiographic findings. Radiographic findings can include fractures of varying ages and stages of healing, Wormian bones (extra bones pieces that occur within a suture of the skull), "codfish" vertebrae (vertebrae with biconcave sides rather than the normal rectangular appearance), and osteopenia. As Case 1 illustrates, the recognition of OI is crucial to differentiating abuse from accidental or pathological trauma. The radiographic findings of abused children include epiphyseal and metaphyseal fractures of the long bones. In contrast, OI fractures usually occur in the shaft of long bones with relative sparing of the metaphysis. Biochemical testing, which involves culturing dermal fibroblasts and analyzing the structure and quantity of type I collagen synthesized and secreted in vitro, has been the standard diagnostic test in the past. However, genomic DNA sequencing is becoming more sensitive than biochemical testing and identifies the specific mutation which can be helpful for evaluation of other family members. Thus, molecular genetic testing is now the preferred diagnostic test in most cases. Family history that is positive for a family member who has frequent fractures, scoliosis, or other orthopedic conditions should raise the suspicion of OI. However, family history may not be completely reliable since there is variability in the expression of the condition even among family members, or the patient’s condition may be due to spontaneous (de novo) mutation.
Management focuses on physical therapy and often includes orthopedic intervention. A careful fracture history is important to target weak bones in an individual and specifically strengthen the muscles around those bones. In addition, children with OI should be seen by a dentist and have a hearing evaluations performed regularly. While no standard medical therapy is currently available, there are many therapies under investigation such as using bisphosphonates to help patients in increasing bone mass and bone strength; human growth hormone to improve linear growth rates and bone formation; and bone marrow transplant to increase the amount of type I collagen.
Marfan syndrome is an autosomal dominant disorder associated with mutation of the FBN1 gene. FBN1 codes for fibrillin-1, an important extracellular matrix protein that is a major constituent of microfibrils and a regulator of TGF-ß (transforming growth factor beta) signaling. Mutations in FBN1 can lead to quantitative or qualitative abnormalities of fibrillin-1 protein. The disease is associated with skeletal, ocular, and cardiovascular manifestations. Intellectual disability is not associated with Marfan syndrome. The diagnosis of Marfan syndrome is based on family history and the documentation of characteristic findings in multiple organ systems. There have been revisions of the diagnostic criteria for Marfan syndrome, with the most recent one in 2010.
In the absence of family history, the diagnosis can be made on the basis of the presence of one of four criteria:
. . . . . (1) aortic root dilation (z-score>2) and ectopia lentis (dislocated lens)
. . . . . (2) aortic root dilation with an FBN1 mutation
. . . . . (3) aortic root dilation with sufficient system findings (a systemic score >7)
. . . . . (4) ectopia lentis with an FBN1 mutation with known associated aortic root dilation
In the presence of family history, the diagnosis can be made in the presence of one of three criteria:
. . . . . (1) ectopia lentis,
. . . . . (2) systemic findings (a systemic score >7)
. . . . . (3) aortic root dilation
Systemic findings include long arms, legs, and fingers (archnodactyly), tall and thin body type, scoliosis, chest deformity, flexible joints, flat feet, crowded teeth, and skin stretch marks. A "systemic score" can be calculated as follows:
. . . . . Wrist sign (+1 point): The tip of the thumb covers the entire nail of the fifth finger when wrapped around the contralateral wrist.
. . . . . Thumb sign (+1 point): The entire distal phalanx of the adducted thumb extends beyond the ulnar border of the palm.
. . . . . Both wrist and thumb signs are present (+3 points instead of the +1 point for each).
. . . . . Pectus carinatum (+2 points)
. . . . . Pectus excavatum or chest asymmetry (+1 point)
. . . . . Hindfoot valgus deformity (+2 points)
. . . . . Flat feet (Pes planus) (+1 point)
. . . . . Spontaneous pneumothorax (+2 points)
. . . . . Lumbosacral dural ectasia (+2 points): Widening or ballooning of the dural sac surrounding the spinal cord.
. . . . . Protrusio actabulae: (+2 points): Medial bulging of the acetabulum into the pelvis.
. . . . . Scoliosis or thoracolumbar kyphosis (+1 point)
. . . . . Reduced elbow extension (+1 point)
. . . . . Facial features (+1 point): Must have at least 3 of the following 5: 1) Dolichocephaly (disproportionately long and narrow head), 2) Downward slanting palpebral fissures, 3) Enophthalmos (recenssion of the eyeball within the orbit), 4) Retrognathia (jaws recede with respect to the frontal plane of the forehead), 5) Malar hypoplasia (underdeveloped cheekbones).
. . . . . Skin striae (+1 point): These resemble stretch marks but are not associated with stretching forces.
. . . . . Severe myopia (+1 point)
. . . . . Mitral valve prolapse (+1 point): Defined by echocardiography.
. . . . . Reduced upper segment to lower segment and increased arm span to height ratios (requires calculation).
As discussed in Case 2, spontaneous pneumothorax should raise suspicion for hereditary connective tissue disorders including Marfan syndrome. Lung bullae can develop in patients with Marfan syndrome, especially of the upper lobes, and can predispose to spontaneous pneumothorax.
Life expectancy is reduced in patients with Marfan syndrome, commonly due to progressive dilation of the aortic root and an increased risk of aortic dissection with advancing age. Death often occurs in the third decade in the absence of palliative surgery to prevent aortic valvular regurgitation and aortic rupture. The management of aortic aneurysm in patients with Marfan syndrome often includes administration of antihypertensive such as a beta-blocker. Surgical repair of the aorta is indicated when the maximal measurement approaches 5 cm in adults or older children, the rate of increase of the aortic diameter approaches 1 cm per year, or progressive aortic regurgitation occurs.
Ehlers-Danlos Syndrome (EDS) is a group of inherited disorders caused by quantitative or qualitative defects of various types of fibrillar collagen. The result is a wide clinical spectrum of diseases with various features that can include skin hyperelasticity, fragility of the skin, and blood vessels, delayed wound healing, and joint hypermobility. Clinical presentation usually occurs after birth. The defect can occur at any step in the production of collagen fibers. The procollagen fiber itself can be defective, as can enzymes that perform the post-translational hydroxylation of lysine, or any of the enzymes which chaperone the assembly of procollagen fibers into normal collagen. EDS has been reclassified into the clinical forms described in the table below.
Skin fragility in EDS is caused by a thin dermis depleted of collagen fibrils. Its texture is often described as soft and velvety. Tears or lacerations over bony prominences commonly occur. Minor trauma often cause large gaping wounds which heal slowly. Surgery can be complicated by wound dehiscence. Patients will often present to ERs with a history of multiple lacerations requiring suture closure despite relatively minor trauma. Vascular fragility may be due to defects in collagen type 3, resulting in vessels with low tensile strength. In these individuals, aneurysms, arteriovenous malformations, and vascular dissections are common. Ecchymoses are present with hemosiderin deposits over bony prominences. Hyperextensibility and joint hypermobility are caused by ligamentous laxity (which predisposes to dislocated hips in infants). Clubfoot, joint effusions, and spondylolisthesis (vertebral displacement) may also be present. The gastrointestinal tract can be similarly affected due to the reduced tensile strength of the bowel walls that predisposes to spontaneous rupture. Bony involvement usually manifests as kyphosis. Individuals diagnosed with EDS usually display one or a combination of these different symptoms. Life expectancy is highly variable in EDS as demonstrated in the different clinical forms. The most severe complications of disease result from bowel and vascular weakness. No curative therapy is available.
Clinical forms of Ehlers Danlos Syndromes
| Brief Description | ||
|---|---|---|
| Characterized by premature birth caused by rupture of membranes, skin hyperelasticity and fragility, easy bruising, generalized and severe joint hypermobility, scoliosis, and mitral valve prolapse. Life expectancy is not reduced | ||
| Characterized by severe joint hypermobility and minimal skin manifestation | ||
| Characterized by most pronounced dermal thinning. Patients with this vascular form have a shortened life span and increased morbidity due to premature birth, extensive ecchymoses from trauma, and rupture of the bowel, uterus, or medium sized arteries | ||
| Characterized by joint hyperextensibility, hypotonia, kyphoscoliosis, fragile cornea, keratoconus, skin hyperelasticity, and fragile bones | ||
| Characterized by short stature, marked joint hyperextensibility and dislocation | ||
| Characterized skin hyperelasticity and marked joint hypermobility | ||
| Characterized by premature rupture of membranes, delayed closure of fontanels, skin fragility and laxity, easy bruisability, growth retardation, short limbs, umbilical hernia, and characteristic facies |
Other connective tissue disorders to consider in the differential diagnosis, depending on the presenting feature include homocystinuria and Stickler syndrome. Homocystinuria is often discussed with Marfan syndrome since there are significant similarities. It is an inborn error of methionine metabolism characterized by variable intellectual disability, ectopia lentis and/or severe myopia, skeletal abnormalities, and a tendency for intravascular thrombosis and thromboembolic events. Distinguishing features are upward lens dislocation in homocytinuria compared to downward lens dislocation in Marfan syndrome, and intellectual disability in homocystinuria compared to normal intellect in Marfan syndrome. Stickler syndrome is characterized ocular findings such as severe myopia, sensorineural and/or conductive hearing loss, hypermobility, early osteoarthritis, cleft palate, micrognathia (Pierre Robin sequence) and other distinctive facial features.
Questions:
1. How is osteogenesis imperfecta differentiated from child abuse?
2. How are future fractures prevented in children with OI?
3. What are the main organ systems that are affected in patients with Marfan syndrome?
4. What is the most common cause of early death in individuals with Marfan syndrome?
5. What are 3 of the cardinal features of Ehlers-Danlos?
6. How is homocystinuria differentiated from Marfan syndrome clinically?
Related x-rays
Below: This is an occult osteogenesis imperfecta case in an infant presenting with a femur fracture due to minor trauma from: Yamamoto LG. Fussiness Following Minor Trauma in an Infant. In: Yamamoto LG, Inaba AS, DiMauro R (eds). Radiology Cases In Pediatric Emergency Medicine, 1999, volume 6, case 2. Read the online case and description at: www.hawaii.edu/medicine/pediatrics/pemxray/v6c02.html
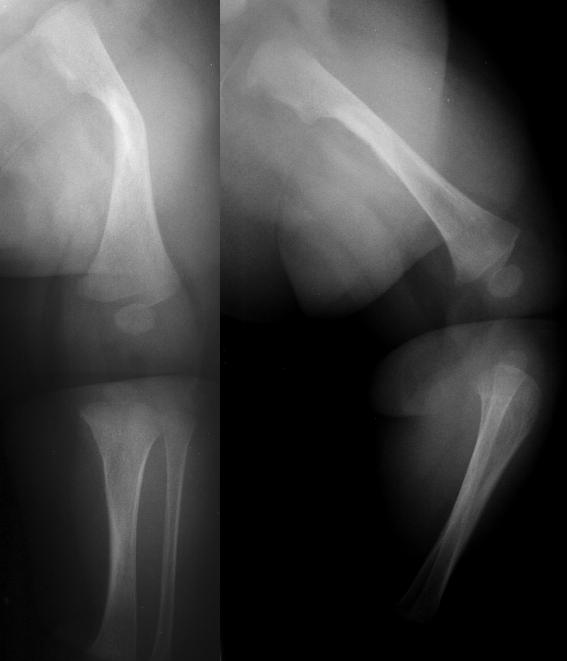
Below: This is a more severe osteogenesis imperfecta case from: Yamamoto LG. Vomiting and Coughing in a 3-Month Old With Weak Bones. In: Yamamoto LG, Inaba AS, DiMauro R (eds). Radiology Cases In Pediatric Emergency Medicine, 1999, volume 6, case 3. Read the online case and description at: www.hawaii.edu/medicine/pediatrics/pemxray/v6c03.html
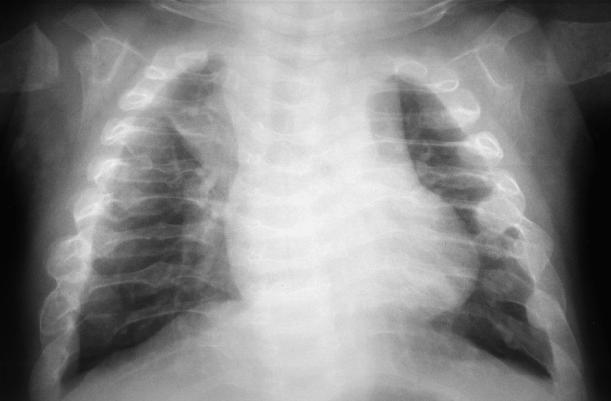
This chest X-ray shows multiple rib fractures and osteopenia.
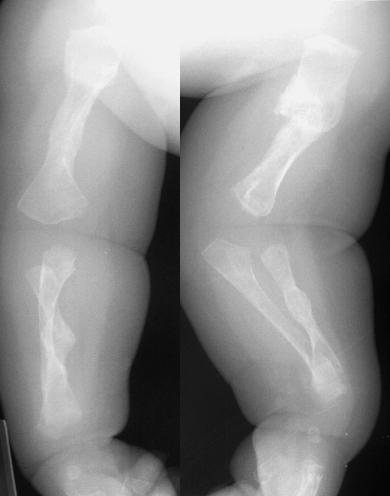
This X-ray of both upper extremities shows multiple fractures and crumpling of the long bones.
Below: This teen presents with an acute aortic dissection from: Feng AK. Severe Acute Chest Pain in an Adolescent. In: Yamamoto LG, Inaba AS, DiMauro R (eds). Radiology Cases In Pediatric Emergency Medicine, 1995, volume 3, case 12. Read the online case, description, and view the other radiographs at: www.hawaii.edu/medicine/pediatrics/pemxray/v3c12.html
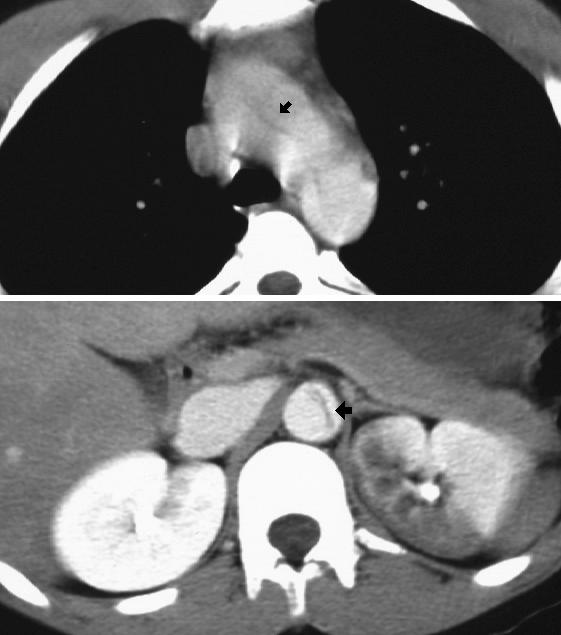
The black arrow in the upper image shows the intimal flap within the aortic arch. The black arrow in the lower image shows the intimal dissection in the descending aorta. Part of the left kidney is infarcting suggesting that the dissection encompasses part of the left renal arter.
References
1. Bolar N et al. Marfan syndrome: from gene to therapy. Curr Opin Pediatr 2012;24(4):498-504.
2. Cundy T. Recent advances in osteogenesis imperfecta. Calcif Tissue Int 2012;90(6):439-449.
3. Dietz H. Marfan syndrome. In: Pagon RA et al. GeneReviews. http://www.ncbi.nlm.nih.gov/books/NBK1335/ Initial Posting: April 18, 2001; Last Update: December 1, 2011. Accessed 2/25/13.
4. Morelli JG. Chapter 651: Disease of the Dermis. Ehlers Danlos Syndrome. In: Kliegman RM et al. Nelson Textbook of Pediatrics, 19th edition. 2011. Philadephia: W. B. Saunders Company, pp. 2278.
5. Royce PM, Steinmann B (eds). Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects 2nd edition. 2002. New York: John Wiley & Sons.
6. Pyeritz RE. The Marfan syndrome. Ann Rev Med 2000;51:481-510.
7. Shapiro JR and Sponsellor PD. Osteogenesis imperfecta: questions and answers. Curr Opin Pediatr 2009;21(6):709-716.
8. Steiner R et al. COL1A1/2-Related Osteogenesis ImperfectaIn: Pagon RA et al. GeneReviews. http://www.ncbi.nlm.nih.gov/books/NBK1295/ Initial Posting: January 28, 2005; Last Update: February 14, 2013. Accessed 2/25/13.
9. Tosi L. Osteogenesis imperfecta. Curr Opin Pediatr 1997;9:94-99.
Answers to questions
1. These can be difficult to distinguish. Factors that can help distinguish these two include the presence of associated physical findings, family history, location of fracture (femur and radius vs. tibia and radius), type of fracture (comminuted mid shaft vs. epiphyseal and greenstick), radiographic appearance of the fractures (i.e., presence of osteopenia).
2. Instruction in safe handling for caregivers, identifying weak bones, bracing, physical therapy.
3. Skeletal, ocular, and cardiovascular systems.
4. Aortic root dilation causing aneurysm and dissection.
5. Any three of the following: hyperextensible velvety skin, atrophic scars, joint hypermobility, connective tissue fragility, and bruising.
6. Marfan syndrome, unlike homocystinuria, is not associated with intellectual disability. Homocystinuria associated with venous thrombosis and downward dislocation of the ocular lens (vs. upward dislocation in Marfan syndrome).