This is a 12-year-old male who presents to the ED with complaints of palpitations for the past 3 days. He reports feeling episodes of palpitations occurring up to 10 times per day, lasting between 5 to 30 minutes. There have been no associated syncopal episodes. His caffeine intake is minimal and is taking no regular medications. His past medical history reveals three generalized tonic-clonic seizures during the past 2 years.
Exam: T 37.7, P 90, R 20, BP 115/72, height 140 cm (10%ile), weight 41 kg (50%ile). Alert and in no distress. He has rounded facies with a stocky build. HEENT, neck, heart, lungs and abdomen exams are normal. His hands show brachydactyly. Tanner II pubertal development. Neurologic exam reveals symmetric hyperreflexia and a positive Chovstek sign. Inflation of a sphygmomanometer induces a Trousseau sign.
Laboratory studies: Na 141 mEq/L, K 4.2 mEq/L, Cl 100 mEq/L, bicarbonate 25 mEq/L, BUN 8 mg/dL, creatinine 0.45 mg/dL, glucose 91 mg/dL, calcium 6.0 mg/dL (normal 8.5 to 10.5), ionized calcium 0.65 mmol/L (normal 1.00 to 1.30), phosphorus 10.6 mg/dL (normal 3.5 to 6), magnesium 1.8 mg/dL (1.5-2.3), alkaline phosphatase 1070 U/L (normal 125 to 450), PTH 750 pg/mL (10-65). An electrocardiogram revealed frequent ventricular extrasystoles with mild QT prolongation (QTc: 0.47 sec). Radiographs of his hands show short 3rd to 5th phalangeal and metacarpal bones. Computed tomography of his brain demonstrates small basal ganglia calcifications.
The phenotypic stigmata of Albright Hereditary Osteodystrophy and characteristic laboratory findings result in a diagnosis of Pseudohypoparathyroidism type 1a. He is admitted to the hospital and is treated with parenteral 10% calcium gluconate for three days (10 mL/dose). The ventricular extrasystoles disappeared and his QT interval normalized soon after intravenous calcium administration. He is discharged on 1.0 mcg/day of calcitriol (1,25-dihydroxyvitamin D3), 1000 mg elemental calcium daily, and a low-phosphorus diet.
Calcium is an essential component of the mineral portion of bone and as its divalent cation (Ca++) is necessary for the function of every cell. The skeleton contains 98% of total body calcium. The remaining 2% circulates, of which 50% is bound to protein, mainly albumin, and the other 50% is ionized or free calcium, which exerts the physiological effects (intracellular communication, interneuronal transmission, muscle contraction, clotting, cellular proliferation, synthesis and secretion of endocrine and exocrine factors, enzyme cofactor). The plasma calcium level is maintained by the interplay of three dynamic processes: tubular reabsorption from the kidneys, absorption from the small intestine and bone remodeling. The two main calciotropic hormones that influence these processes through feedback-loop mechanisms are parathyroid hormone (PTH) and 1,25-dihydroxyvitamin-D3. The calcium sensing receptor (CaSR) is also a critical regulator of plasma calcium levels by directly influencing PTH release in response to circulating calcium levels.
PTH acts to mobilize calcium from bone by increasing osteoclast activity through an action on osteoblasts, and acts on the kidney to increase reabsorption of calcium in the distal renal tubule, enhance 1-alpha hydroxylation of 25-hydroxyvitamin-D to 1,25-dihydroxyvitamin-D3, to increase calcium absorption from the small intestine, and increase renal tubular phosphate excretion.
1,25-Dihydroxyvitamin-D3 is the active metabolite of vitamin D. It promotes calcium and phosphate absorption from the intestine, increases bone mineralization and increases calcium reabsorption in the distal tubule of the kidney. When dietary calcium intake or serum calcium concentration is low, 1,25-dihydroxyvitamin-D3 interacts with the vitamin D receptor in osteoblasts to induce bone resorption, releasing calcium into the circulation.
The CaSR is a G-protein coupled membrane receptor that regulates calcium levels through its expression in the parathyroid gland and renal tubule. Activation of the CaSR from increased ionized calcium leads to inhibition of PTH secretion and increased renal calcium excretion.
Etiologies of Hypocalcemia in Children and Adolescents
1) Hypoparathyroidism
. . . a) Congenital
. . . . . . Transient neonatal
. . . . . . Dysgenesis / agenesis of parathyroid glands
. . . . . . . . . Isolated hypoparathyroidism
. . . . . . . . . Hypercalciuric hypocalcemia
. . . . . . . . . DiGeorge syndrome (del 22q11.2)
. . . . . . . . . Mitochondrial fatty acid disorders (Kearns-Sayre syndrome, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes))
. . . . . . Insensitivity to parathyroid hormone
. . . . . . . . . Pseudohypoparathyroidism, typs Ia, Ib, Ic, II
. . . . . . . . . Pseudopseudohypoparathyroidism
. . . . . . . . . Hypomagnesemia
. . . . . . . . . Dyshormonogenesis
. . . b) Acquired
. . . . . . Autoimmune polyglandular syndrome type I
. . . . . . Activating antibodies to CaSR
. . . . . . Post-surgical, radiation destruction
. . . . . . Infiltrative
. . . . . . . . . excessive iron or copper deposition
. . . . . . . . . granulomatous or neoplastic invasion
. . . . . . Maternal hyperparathyroidism
. . . . . . Hypomagnesemia
2) Vitamin D deficiency
3) Other causes
. . . a) Ca deficiency
. . . b) Hypomagnesemia / Hypermagnesemia
. . . . . . Bartter syndrome
. . . . . . Renal tubular acidosis
. . . . . . Acute renal failure
. . . . . . Chronic inflammatory bowel disease / intestinal resection
. . . c) Hyperphosphatemia
. . . . . . Renal failure
. . . . . . Phosphate administration
. . . . . . Tumor lysis syndrome
. . . . . . Muscle injury (rhadomyolysis, crush)
. . . d) Miscellaneous
. . . . . . Hypoproteinemia
. . . . . . Drugs (furosemide, bisphosphonates, calcitonin, antineoplastic agents, ketoconazole)
. . . . . . Hungry bone syndrome
. . . . . . Acute and critical illness (sepsis, toxic shock, pancreatitis)
The child with hypocalcemia may be asymptomatic and identified through chemistries obtained for another reason or present with intermittent muscular cramping, paresthesias, tetany, carpopedal spasms, laryngospasms, or seizures. Review of symptoms and past medical history may reveal intermittent symptoms associated with hypocalcemia. Commonly, the physical exam is unremarkable other than that of increased neuromuscular irritability: hyperreflexia, Chvostek sign (twitching of the circumoral muscles when tapping lightly over the facial nerve) or Trousseau sign (carpopedal spasm when maintaining a blood pressure cuff 20 mm Hg above the systolic blood pressure for 3 minutes), and occasionally cataracts or abnormal dentition. The physical exam may disclose the characteristic phenotype of Albright hereditary osteodystrophy (pseudohypoparathyroidism type Ia; short stature, round facies, shortened metacarpals, subcutaneous calcification), the DiGeorge syndrome (typical facies, cardiac abnormalities), or rachitic changes (bowed legs or "knock knees", widened metaphyses of the long bones, prominent costochondral junctions, frontal bossing) in the case vitamin D deficiency.
DiGeorge syndrome is usually classified as an immune deficiency, but it usually presents initially with congenital heart disease or with hypocalcemic seizures or tetany. During embryogenesis, the thymus and parathyroid glands originate from the same branchial pouch, which explains why the two abnormalities occur together. Congenital heart disease may be detected during the newborn period. Subsequently, the child will present with hypocalcemic tetany or seizures during the first few months of life, before any opportunistic infection is likely to occur. A chest X-ray will show a cardiac silhouette without the usual thymic shadow. Thymic absence with hypocalcemia is highly indicative of DiGeorge syndrome.
The laboratory evaluation of a hypocalcemic child requires measuring serum total and ionized calcium, PTH, magnesium, phosphate, creatinine, alkaline phosphatase, and urinary calcium. If there are concerns of a metabolic bone disease, serum levels of 25-hydroxyvitamin-D and 1,25-dihydroxyvitamin-D should also be obtained.
Calcium and magnesium tend to be antagonistic, so hypocalcemia will occur in association with a high magnesium load, which most commonly occurs in premature neonates of mothers treated with magnesium tocolytics. Severe hypomagnesemia will also result in hypocalcemia since magnesium is a required co-factor for PTH release. Thus, both extremes of magnesium, will result in hypocalcemia.
A patient with hypocalcemia, hypocalciuria, hyperphosphatemia, and low serum PTH levels has hypoparathyroidism caused by a primary defect in PTH synthesis or secretion. An elevated PTH level indicates a compensatory increase in response to hypocalcemia or a resistance to PTH (pseudohypoparathyroidism). Vitamin D deficiency can also present with hypocalcemia and an elevated PTH, mimicking pseudohypoparathyroidism.
Management of symptomatic acute hypocalcemia is intravenous 10% calcium gluconate (93 mg elemental Ca in 10 mL), 1-2 mL/kg over 10 minutes. Although calcium chloride can also be used, some texts discourage the use of calcium chloride because it can lead to metabolic acidosis. After acute symptoms have resolved, an intravenous infusion of calcium should be initiated at a rate to keep the serum calcium levels in the low normal range while an investigation of the etiology ensues. Therapy for patients with hypo- or pseudohypoparathyroidism is individualized using calcitriol (20-60 ng/kg/day) and supplemental oral calcium (30-75 mg elemental Ca/kg/day). Frequent measurements of serum calcium and creatinine and renal ultrasonography are done to monitor for hypercalcemia, hypercalciuria, and nephrocalcinosis.
Etiologies of Hypercalcemia in Children and Adolescents
1)Hyperparathyroidism
. . . a) Sporadic
. . . b) Familial
. . . . . . Neonatal severe hyperparathyroidism (NSHPT)
. . . . . . Multiple endocrine neoplasia, type I
. . . . . . Multiple endocrine neoplasia, type IIa
. . . . . . McCune-Albright syndrome
. . . . . . Familial hyperparathyroidism-jaw tumor syndrome
. . . . . . Jansen's metaphyseal dysplasia
. . . c) Secondary / Tertiary
. . . . . . Postrenal transplantation
. . . . . . Chronic hyperphosphatemia
. . . d) Ectopic production of PTHrP (parathyroid hormone-related protein)
2) Familial Hypocalciuric Hypercalcemia (FHH)
. . . a) Loss-of-function mutation in CaSR
. . . b) Inhibitory autoantibodies to the calcium-sensing receptor
3) Excessive intake of calcium or vitamin D
. . . a) Nutritional milk-alkali syndrome
. . . b) Exogenous ingestion (vitamin D) or topical application (calcitriol or analog)
. . . c) Ectopic production of calcitriol associated with granulomatous diseases: sarcoidosis, inflammatory bowel disease, TB, histoplasmosis, coccidioidomycosis, leprosy, HIV, lymphoma, dysgerminoma
4) Immobilization
5) Other causes
. . . a) Williams-Beuren syndrome (del 7q11.23)
. . . b) Neoplasia: osseous metastases, production of PTHrP, cytokines/osteoclast activation
. . . c) Hypophosphatasia
. . . d) Drugs: thiazides, diuretics, lithium, vitamin A and analogs, calcium, alkali, anti-estrogens
. . . e) Total parental nutrition
. . . f) Endocrinopathies: hyperthyroidism, hypoadrenocorticism, pheochromocytoma
. . . g) Vasoactive intestinal polypeptide-secreting tumor
. . . h) Acute or chronic renal failure / administration of aluminum
. . . i) Juvenile idiopathic arthritis, cytokine medicated
Clinical features are dependent on the underlying disorder and degree of hypercalcemia. Nonspecific symptoms include polydipsia, polyuria, anorexia, constipation, nausea, vomiting, abdominal pain, weakness, and altered consciousness. The patient may show signs of dehydration or altered mental status, otherwise the physical exam is usually normal. Commonly a shortened QT interval can be found on EKG and nephrocalcinosis and/or renal calculi can be demonstrated by ultrasonography.
Primary hyperparathyroidism is much less common in children and adolescents than in adults, is usually sporadic, and is nearly always due to a single parathyroid adenoma. Nearly all patients are symptomatic at presentation and end-organ damage (nephrocalcinosis, nephrolithiasis, acute pancreatitis, or bone involvement) is common.
Primary hyperparathyroidism can also be an autosomal dominant genetic disorder that is typically associated with multi-gland hyperplasia and is most commonly the presenting manifestation of multiple endocrine neoplasia type I.
Young children and infants may develop vitamin D intoxication when receiving only 2000 to 4000 units daily. Serum levels of 25(OH)D are elevated, but serum levels of calcitriol are usually normal and PTH levels are suppressed.
Endogenous vitamin D intoxication can occur with granulomatous disease and other inflammatory disorders. Infectious diseases such as cat scratch fever, coccidioidomycosis, histoplasmosis, leprosy, and TB have all been associated with hypercalcemia in children. In these conditions, activated T cells and macrophages express 25-(OH)D-1-alpha hydroxylase activity, which converts 25-hydroxyvitamin-D to calcitriol.
Immobilization is a common cause of hypercalciuria in children and adolescents and can also cause hypercalcemia. When a rapidly growing child is immobilized or placed on bed rest, there is a marked decrease in osteoblastic bone formation and a corresponding increase in osteoclastic bone resorption. This imbalance in bone remodeling leads to excessive mobilization of calcium (and phosphate) from the skeleton.
Hypercalcemia occurs in fewer than 1% of children with cancer, and has been reported with leukemia, lymphoma, myeloma, neuroblastoma, hepatocellular carcinoma, ovarian carcinoma, hepatoblastoma, rhabdomyosarcoma, brain cancer, and dysgerminomas. Malignancy-associated hypercalcemia can be attributed to two general mechanisms: osteolytic, due to direct invasion of the skeleton by tumor cells, and humoral, due to tumor production of circulating factors that activate osteoclastic bone resorption.
The most commonly identified humoral factor that causes hypercalcemia of malignancy is PTHrP (paraghyroid hormone-related protein). Some tumors secrete sufficient PTHrP into the circulation to induce hypercalcemia via interaction with the type 1 PTH/PTHrP receptor. Other tumor-produced factors that play a role in producing hypercalcemia include calcitriol, prostaglandins, interleukin-1 and interleukin-6, transforming growth factor-b, and tumor necrosis factor.
The initial laboratory evaluation of a hypercalcemic child requires measuring serum total and ionized calcium, urinary fractional excretion of calcium, PTH, phosphate, creatinine, 25-hydroxyvitamin-D and 1,25-dihydroxyvitamin-D levels.
In the absence of secondary hyperparathyroidism (chronic renal failure, ingestion of thiazides or lithium), consistently elevated PTH levels are indicative of primary hyperparathyroidism. 25-hydroxvitamin-D levels are increased in patients with hypercalcemia caused by excessive intake of vitamin D. 1,25-dihydroxyvitamin-D levels are increased in patients with granulomatous, chronic inflammatory, and lymphomatous diseases or those receiving that vitamin. PTHrP levels are high in children with humoral hypercalcemia of malignancy. Children with asymptomatic, mild hypercalcemia, and decreased fractional excretion of calcium likely have FHH (familial hypocalciuric hypercalcemia).
Appropriate management depends on the severity and cause of the high calcium levels. If the calcium level is less than 12 mg/dL and the patient is asymptomatic, treatment may be delayed pending evaluation of the cause. Symptomatic patients and those with calcium levels greater than 12 mg/dL should be treated because of the adverse effects of hypercalcemia on the cardiac, renal, gastrointestinal, and central nervous systems.
The initial approach to the medical treatment of severe or symptomatic hypercalcemia is to increase the urinary excretion of calcium. Normal saline containing 30mEq/L KCl should be infused to correct dehydration and maximize the glomerular filtration rate. Furosemide and other powerful loop diuretics are rarely necessary, and can induce excessive diuresis and dehydration; the consequent fall in the glomerular filtration rate can worsen hypercalcemia.
In most cases, hypercalcemia is due to osteoclastic bone resorption, and agents that inhibit or destroy osteoclasts will be effective treatments. Calcitonin (2 to 4 U/kg per 12 h) given by subcutaneous injection is effective at first, but resistance to the hormone develops rapidly. Bisphosphonates (pamidronate, etidronate, zoledronic acid) induce osteoclast apoptosis and are potent inhibitors of bone resorption. They can rapidly lower serum and urinary calcium levels in patients with hypercalcemia due to a variety of causes, and the effects can last for weeks. Patients must be monitored carefully, as these powerful agents can cause severe hypocalcemia, hypophosphatemia, and hypomagnesemia. Because of concerns regarding potential late adverse effects of bisphosphonates on growth and development of the skeleton, calcitonin tends to be used more frequently in children because it has no long-term sequelae.
Parathyroid surgery is recommended for all children with primary hyperparathyroidism. Hypercalcemic children with FHH will rarely require any intervention. The secondary hyperparathyroidism of chronic renal failure is best treated by lowering the serum phosphate to the extent possible while maintaining the serum calcium level in the low-normal range with calcitriol (1,25-dihydroxyvitamin-D).
Glucocorticoids are effective in lowering excess calcium levels due to vitamin D ingestion, granulomatous and inflammatory diseases, or malignancies, by inhibiting renal 25-hydroxyvitamin-D-1-alpha-hydroxylase activity; however, they are used with caution in children because they impair linear growth and bone mineralization.
Children with Williams syndrome or idiopathic infantile hypercalcemia often have mildly elevated serum levels of calcitriol, and a low calcium formula in the infant or reduced calcium diet in the older child may be all that is needed to treat the hypercalcemia and hypercalciuria.
A low calcium diet, copious fluids, avoidance of vitamin D, and early mobilization are indicated in the immobilized child to avoid hypercalcemia. Bisphosphonate treatment has also been used.
Questions
1. What are the effects of PTH on the kidney?
2. True / False: Elevated levels of parathyroid hormone always result in hypercalcemia.
3. Inactivation mutations of the Calcium Sensing Receptor (CaSR) results in what condition?
References
Kelly A, Levine MA. Disorders of calcium, phosphate, parathyroid hormone and vitamin D. In: Kappy MS, Allen DB, Geffner ME (eds). Pediatric practice: endocrinology. 2009, Springfield: Charles C. Thomas Publisher, Ltd, pp. 191-256.
Lietman SA, Germain-Lee EL, Levine MA. Hypercalcemia in children and adolescents. Curr Opin Pediatr 2010;22:508-515.
Root AW, Diamond FD. Disorders of Mineral Homeostatsis in the Newborn, Child, and Adolescent. In: Sperling MA (ed). Pediatric endocrinology. 3rd ed. 2008, Philadelphia: Saunders, pp 686-769.
Zhou P, Markowitz M. Hypocalcemia in Infants and Children. Pediatr Rev 2009;30:190-192.
Related x-rays
DiGeorge Syndrome:
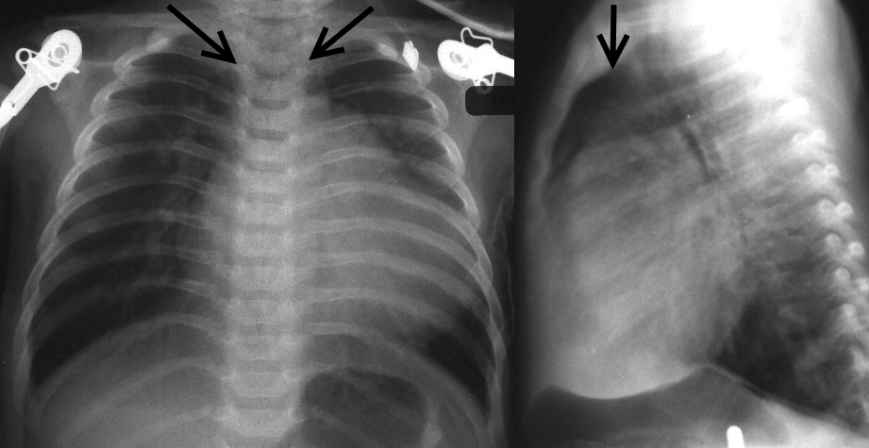
The arrows point to the mediastinum in this 2-month old who should have a normal large thymus occupying this space which is not present. The heart size is large because of a VSD. The patient presents with seizures due to hypocalcemic tetany due to DiGeorge syndrome.
Yamamoto LG. Seizure and VSD in a 2-month Old Infant. In: Yamamoto LG, Inaba AS, DiMauro R. Radiology Cases In Pediatric Emergency Medicine, 1995, volume 2, case 2. Review the case and x-ray images online at: www.hawaii.edu/medicine/pediatrics/pemxray/v2c02.html
Rickets:
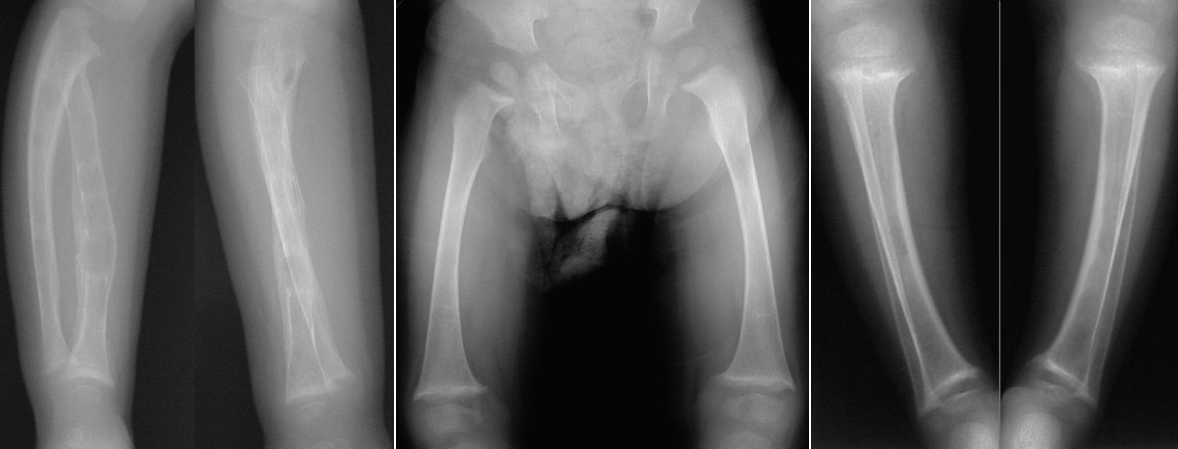
The left image shows both forearms which are demineralized. The middle image shows bowing of both femurs. The right image shows bowing and demineralization of the tibiae/fibulae.
Mendez D. Elbow Swelling In a 2 Year Old With Liver Disease. In: Yamamoto LG, Inaba AS, DiMauro R. Radiology Cases In Pediatric Emergency Medicine, 1999, volume 6, case 5. Review the case and x-ray images online at: www.hawaii.edu/medicine/pediatrics/pemxray/v6c05.html
Answers to questions
1. Increased calcium absorption, increased phosphate excretion, and increased 25-hydroxy-vitamin-D-1-alpha-hydroxylation (increased calcitriol production).
2. False. High PTH levels should result in hypercalcemia; however, pseudohypoparathyroidism is an end-organ resistance to PTH, so despite an elevated PTH, patients have hypocalcemia. Hypocalcemia seen in Vitamin D deficiency can also result in elevated PTH levels.
3. Familial hypocalciuric hypercalcemia
Return to Table of Contents
University of Hawaii Department of Pediatrics Home Page
